








Analysis of the difference between PCR, qPCR, RT-PCR and Real-time PCR
 source:ELK Biotechnology
source:ELK Biotechnology date:2023-08-09
date:2023-08-09 views:373
views:373
Introduction of PCR technology
Polymerase chain reaction (polymerase chain reaction, PCR) is a technique for enzymatic amplification of specific DNA fragments in vitro, invented by Kary Mullis and colleagues in 1985. The application of thermostable Taq DNA polymerase and the invention and improvement of automated devices have brought PCR technology into a practical stage. The invention and wide application of this technology have greatly promoted the development of various related disciplines of molecular biology, and the inventor also won the Nobel Prize in Chemistry in 1993.
With the maturity of PCR technology, many related technologies have been derived. This type of technology has the advantages of sensitivity, specificity, rapidity and simplicity. It is more and more widely used in microbial inspection, and many countries have included it in the standard method of inspection; our country's inspection and quarantine field is also expanding its application. The Centers for Disease Control and Prevention above the level have PCR detection technology capabilities, which are used for rapid detection of public health emergencies and effective supervision of food safety.
The basic principle of PCR technology is similar to the natural replication process of DNA in vivo. It is a process of repeated DNA template melting, primer and template DNA binding, and DNA polymerase catalysis to form a new DNA chain in vitro, but the whole process is carried out in vitro, and the reaction The system is simpler than in body.
1.System composition and basic process
1)System composition Classical PCR uses DNA as the template, and the system composition includes the original template, primers, raw materials, DNA polymerase, and appropriate salts, buffers, and temperature cycling parameters. The original template refers to the long double-stranded DNA target fragment to be amplified, which is often the DNA in the sample to be tested in microbial testing. The primers are two single-stranded oligonucleotides whose sequences are identical and complementary to the two ends of the fragment to be amplified, respectively. The raw material is 4 kinds of deoxynucleoside triphosphates.
2)Basic process The basic process of PCR is divided into three stages: denaturation, annealing and extension, and the amplification of the target fragment is achieved after multiple cycles. Denaturation refers to the process of heating the reaction system mixture to 93°C~95°C for a short time, generally 30s, to denature the double-stranded DNA to be tested under the action of high temperature, and melt into two single-stranded DNA templates. Annealing refers to cooling the reaction system mixture to a specific temperature (also called the annealing temperature). Extension refers to the synthesis of two new DNA strands complementary to two template DNA by raising the temperature of the reaction system to 72 ℃ and using four kinds of deoxynucleoside triphosphate as raw materials according to the principle of base pairing under the action of heat-resistant DNA polymerase.
2.The remarkable characteristics of PCR technology determine that it has a very important position in both biological research and microbial testing.
1)High sensitivity: The amount of product increases exponentially, which can amplify the initial template to be tested in the order of picograms (pg=10-12) to the level of micrograms (μg=10-6). A target cell is detected in the cell, and so on.
2)High specificity: Based on the principle of base pairing, the primers can be specifically and correctly combined with the template DNA, the DNA polymerase synthesis reaction has a high degree of fidelity, and the target gene DNA has a high degree of specificity and conservation.
3)Simple and fast process: the reaction solution is added at one time, denaturation-annealing-extension is performed in the DNA amplifier, and the amplification reaction is completed in 2-4 hours; and the amplification product is easy to analyze, free from radioactive contamination, and easy to popularize. In particular, the invention and promotion of real-time fluorescent quantitative PCR can monitor the reaction process in real time through the computer, and no need to detect the product, which greatly shortens the experimental time.
4)Low requirements on sample purity and wide application range: PCR technology has low requirements on sample purity, and crude DNA products can be used as amplification templates; it is not necessary to isolate viruses, bacteria or cultured cells; clinical blood, body cavities can be directly used. Crude DNA amplification detection of liquid, washing liquid, hair, cells, biopsies and other samples. The scope of application is very wide, not only suitable for the rapid detection of microorganisms, but also for clinical and forensic applications.
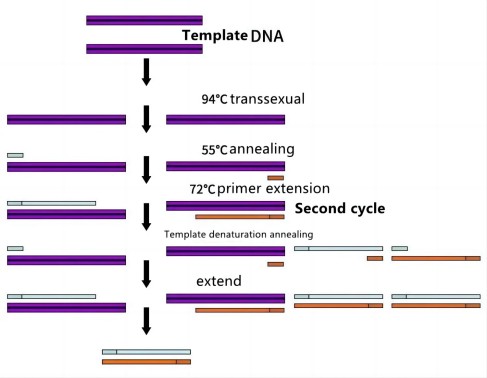
The PCR process can be roughly divided into three steps: denaturation - annealing - extension
Denaturation (90℃-96℃): Under the action of heat, the double-stranded DNA template breaks the hydrogen bonds to form single-stranded DNA.
Annealing: (60℃-65℃): The temperature of the system is lowered, and the primers are combined with the DNA template to form partial double strands.
Extension: (70°C-75°C): Under the action of Taq enzyme (at around 72°C, the best activity), using dNTP as raw material, start from the 3' end of the primer and extend in the direction from 5' to 3' end, Synthesize DNA strands complementary to the template.
As shown in the figure below, after one denaturation, annealing and extension process, the original DNA double-strand with only 1 becomes 2; after 2 times, it becomes 4; A large number of identical DNA fragments are produced.
Follow-up testing
The above is the normal flow of conventional PCR. After the amplification is completed, we need to detect the amplified DNA. Conventional PCR technology usually only uses gel electrophoresis. The electrophoresis technology is not introduced too much here. It is a technology that separates the DNA according to the molecular weight of the DNA, so as to identify and purify the DNA. With the help of gel electrophoresis, it can be judged whether the size of the amplified DNA fragment band is consistent with the size of the target band, so as to know whether the desired product is obtained; see whether there is any heteroband, and judge whether the amplification is specific; see whether there is any Primer-dimers judge the design of primers and so on.
About qPCR and Real-time PCR
Many people mistakenly think that RT-PCR is the abbreviation of Real-time PCR, so they confuse the two, but they are actually different.
In fact, Real-time-PCR and qPCR (Quantitative Rea-time-PCR) are the same thing, both are real-time quantitative PCR, which means that each cycle in the PCR process has a real-time record of data, so the number of starting templates can be determined.
Perform precise analysis.
Although Real-time PCR (real-time fluorescent quantitative PCR) and Reverse transcription PCR (reverse transcription PCR) seem to be abbreviated as RT-PCR, the international convention is: RT-PCR refers specifically to reverse transcription PCR, Real-time PCR is generally abbreviated as qPCR (quantitative real-time PCR). When we just introduced the PCR follow-up detection method, we said that usually we can only use gel electrophoresis to analyze the product, but real-time fluorescence quantitative PCR allows us to detect DNA amplification during the PCR process with the help of fluorescent chemicals. Therefore, there is no need to run electrophoresis, which greatly simplifies the operator's experimental steps.
The experimental principle of qPCR
To put it simply,qPCR is the introduction of fluorescent groups during the entire amplification process. As the number of DNA doubles, the fluorescent signal in the reaction system will also increase. By detecting the intensity of the fluorescent signal, the DNA amplification can be quantitatively analyzed. At present, according to different fluorescent substances, we can roughly divide qPCR into two types. One is to use fluorescent dyes, mainly SYBRGreen I dyes, which can bind to DNA double-strands, and will display fluorescent signals after binding, but almost no fluorescent signals can be detected when there is no binding. enhanced. The other is the use of fluorescent probes, also called TaqMan probe method. The principle is to design a fluorescent probe that can specifically bind to the target gene. The probe contains two groups: a fluorescent group and a A quencher group. Normally, the fluorophore cannot fluoresce due to the existence of the quencher group, but the probe binds to the DNA template during amplification, and two groups are amplified to the probe position. is cleaved so that the fluorophore can fluoresce, and as the copy number increases, the fluorescent signal increases.
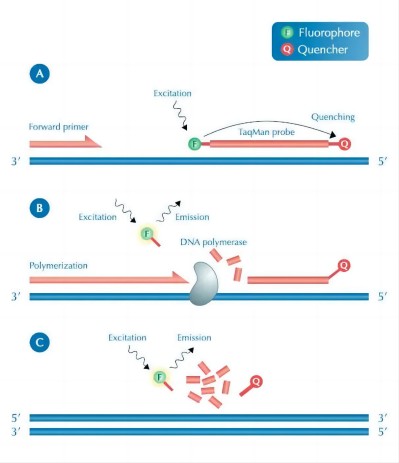
Schematic diagram of the TaqMan probe method
With the help of this quantitative relationship, one can draw the PCR amplification curve, which is the curve of the fluorescence signal changing with the number of cycles. With the help of this curve, the PCR situation can be analyzed. The ideal curve should be presented as " S"-shaped growth, but the content of curve analysis is more complicated, so I won't go into details here for the time being. At least we know that with this technology, we can analyze our PCR results more accurately without having to run electrophoresis every time we finish PCR. Therefore, this technology is more and more widely used. It is also worth mentioning that in the current epidemic situation, how do we determine whether the subject is negative or positive after nucleic acid sampling? The qPCR technology is used. If fluorescence can be detected after a certain number of cycles of amplification Signal, we can think that the subject is positive, otherwise it is negative.
About RT-PCR
RT-PCR is reverse transcription PCR (reverse transcription PCR) or reverse transcription PCR (RT-PCR), which is a widely used variant of polymerase chain reaction (PCR). In RT-PCR, an RNA strand is reverse transcribed into complementary DNA, which is then used as a template for DNA amplification by PCR. First of all, we need to know what reverse transcription is. We generally refer to the process of genetic information flowing from DNA to RNA as transcription. On the contrary, the process of synthesizing DNA using RNA as a template is called reverse transcription. The general process of this technology is to first use RNA as a template to synthesize its corresponding cDNA under the action of reverse transcriptase, and then use the cDNA as a template to perform a common PCR process. In the final analysis, it is to add a reverse transcription process before the PCR process, after which we can use either ordinary PCR methods or qPCR for amplification. For example, viruses usually use RNA as their genetic material. When we detect pathogens, we can only extract its RNA, so reverse transcription is required.
Other PCR techniques
In fact, in addition to the several PCRs introduced above, there are now digital PCR technology, also called absolute quantification of nucleic acid molecules. Compared with fluorescence quantification, this technology is the absolute quantification of DNA number; as well as nested PCR and so on. But its essence is inseparable from the three steps of PCR mentioned at the beginning.

 RETURN
RETURN